How do cancer cells survive self-inflicted damage?
Introduction
A key feature of cancer cells is that their genomes are highly mutated. In other words, compared to normal, healthy cells, cancer cells are fraught with errors in their DNA sequences. This is essential for their evolution, as mutating quickly confers onto them new survival advantages as they replicate and divide along the continuum of tumor initiation to invasion. However, as one may expect, harboring thousands of genomic mutations might not be all beneficial to a cancer cell. After all, these are mistakes in their instruction manual. A nontrivial portion of these errors should likely cause harm.
If cancer genomes are so highly mutated, how do cancer cells survive all their damage?
A fascinating study by Tilk et al [1], appearing in bioRxiv on June 10, 2022, addressed this fundamental question.
Background + intuition
Cancer largely develops via the accretion of somatic mutations after generations of cell division. The vast majority of these mutations are passengers, while drivers — those that actually propel tumor progression — only constitute a small subset. On the other hand, passengers don’t help tumor growth, and might even hinder it.
The role of passenger mutations has long been set aside despite their sheer plenitude, as well as variation, in cancer. Whether the accrual of passengers is truly neutral or instead harmful to the cell has been contested. Some have claimed that passengers have no functional importance, given the observation that most non-synonymous mutations are not selected against in somatic tissues. Conversely, in the germline, negative selection signals are prominent and non-synonymous mutations are expectedly functionally harmful to most genes.
However, why would non-synonymous mutations only be neutral in somatic tissues? This is not convincing, given their known negative consequences in germline cells. Specifically, non-synonymous mutations are damaging in the germline via deleterious effects on protein folding and stability. There is no compelling reason why these negative effects should not be shared between both somatic and germline cells.
In work from last year [2], the authors proposed an explanation for this phenomenon: Hill-Robertson interference. The intuition is that cancer cells divide by mitosis, not recombination; thus, deleterious mutations do not get removed due to genome-wide linkage (also known as Muller’s ratchet). In TCGA tumors, those with very small numbers of passengers (~5% of tumors) show signatures of negative selection, while the remaining majority (>95% of tumors) display signals of inefficient negative selection due to genome-linked mutations. The lack of visible negative selection in most tumors is likely not a reflection of relaxed selective pressures, but instead the ineptitude of selection in removing harmful mutations during mitosis.
Question + hypothesis
So most cancer cells carry mutations that are harmful to their survival. How then does a cancer cell survive a deleterious mutational load? The authors reasonably hypothesized that an accumulation of deleterious mutations in protein-coding regions may destabilize proteins and lead to protein misfolding. This, in turn, may require tumors to regulate their proteostasis: the set of cellular processes that regulate and keep the proteome intact, consisting of protein synthesis, folding, and degradation.
Misfolded proteins can result from non-synonymous/nonsense mutations that cause amino acid substitutions or premature peptide truncations. Thus, protein misfolding may be deleterious to cells not only due to a loss of function incurred in the original protein, but also due to added toxicity resulting from aberrant peptide aggregation in the cell. Thus, the authors state, “is it intriguing to consider the possibility that the need to manage protein misfolding stress is a hallmark of somatic evolution in cancer.”
Genome-wide screen
To address their hypothesis, the authors performed a genome-wide screen to systematically identify which genes are upregulated in their transcription with increased mutational load in ~10,000 tumors in the TCGA. To do this, a generalized linear mixed model (GLMM) was implemented and applied separately to each gene, while accounting for cancer type and tumor purity as confounders. ~5,000 genes emerged from this analysis as upregulated in response to increased mutational load. These genes were subsequently passed into CORUM for a gene set enrichment analysis (GSEA) and gprofiler2 for KEGG pathway enrichment analysis.

Figure 1A in the paper: genome-wide screen framework; generalized linear mixed model applied to each gene in ~10,000 TCGA tumors.
In addition to the expected upregulation of pathways and protein complexes related to the cell cycle, DNA replication, and DNA repair, it was found that terms related to translation, protein degradation, and protein folding were some of the top hits resulting from the enrichment analysis. This was in line with the authors’ hypothesis that tumors bearing high mutational loads experience stress from protein misfolding.
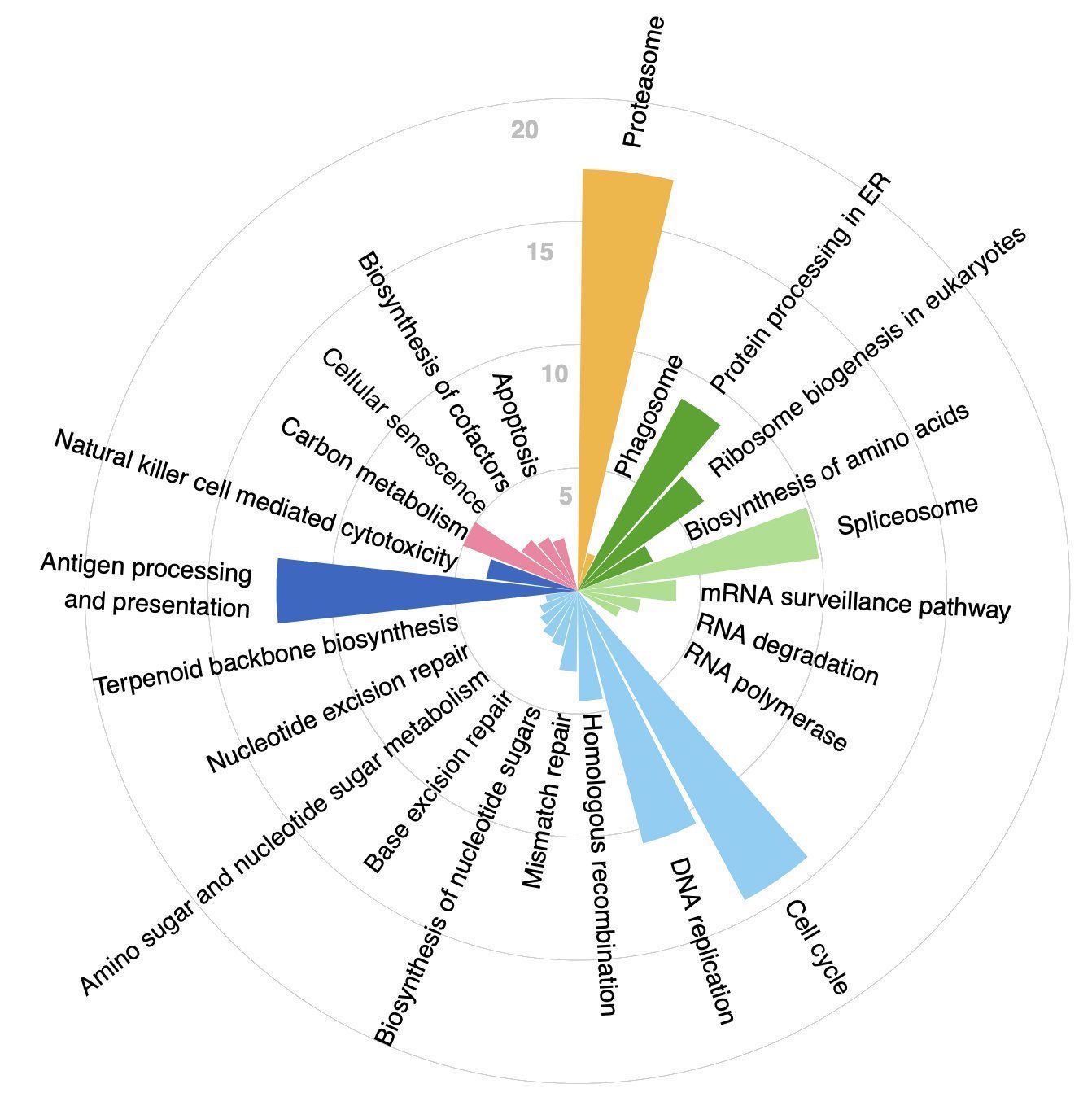
Figure 1C in the paper: enriched pathways resulting from CORUM and KEGG analyses.
Validation in cell lines
Next, the authors inquired whether the transcriptional responses to elevated mutational load seen in TCGA replicated in cancer cell lines from the CCLE. Indeed, they did! This indicated that these responses are generic across highly mutated cancer cells, and cell-autonomous, i.e., independent of external effects such as the immune system, aging, or the microenvironment. The authors claim that this importantly indicates that cancer cell lines are an appropriate model to further causally interrogate effects via various perturbational experiments.

Figure 3 in the paper: cross-validation of TCGA transcriptional responses to high mutational load in cancer cell lines from CCLE.
Causality by knockdowns
To establish causality between proteostasis machinery over-expression and cellular viability under high mutational load, the authors harnessed shRNA knockdown estimates from Project Achilles for the same cell lines from CCLE. To evaluate how mutational load impacts cell viability when protein complexes and gene families underwent loss of function (LOF) via expression knockdown, a simple generalized linear model (GLM) was harnessed, with its beta coefficients reflecting the direction and strength of associations. As expected, the viability in high mutational load cell lines decreased with disruption of proteostasis machinery. Specifically, knockdown of the 19S regulatory particle (protein import machinery) of the proteasome produced significant and robust associations across all cancer types, but knockdown of the 20S core (protein cleaving machinery) was not associated with decreased viability.
Additionally, expression knockdown of mitochondrial ribosomes significantly and robustly decreased viability with increased mutational load in every cancer type, and this was concordant with TCGA expression patterns. Intriguingly, mutational load conversely increased viability in every cancer type when cytoplasmic ribosomes underwent knockdown. In line with the previous analyses, cytoplasmic ribosomes were observed to be down-regulated in response to mutational load in TCGA.

Figure 4 in the paper: causal perturbation experiments in Achilles and PRISM showing beta coefficients of association between knockdown/drug inhibition and cell viability, across all major cancer types.
PRISM drug screen
Appropriately arguing that functional redundancy in the genome leads to noisy expression-knock down estimates, the researchers further studied how drugs targeting the function of entire complexes impact viability with increased mutational load across all cancer types. To do so, they harnessed data from the PRISM drug screen within CCLE. They used a simple GLM again to measure the association between mutational load and cell viability following drug inhibition. Indeed, treatment with the majority of proteasome inhibitors was significantly associated with a decrease in cell viability in cell lines harboring high mutational loads.
The authors noted that the selection of currently available proteostasis drugs in PRISM was limited, but they anticipate that drugs inhibiting other chaperone machinery or splicing complexes could be effective against cancer cells with high mutational burdens.
Taken together, these results beautifully and elegantly demonstrate that elevated expression of protein degradation and folding machinery is causally related to cell viability in highly mutated cell lines — and plausibly in highly mutated tumors. Cells bearing high mutational loads must up-regulate these complexes or they cannot survive, and are vulnerable to disruption of said complexes via shRNA knockdown or targeted therapies.
One final important analysis performed using PRISM data was asking whether high mutational load cancer cell lines are sensitive to other classes of drugs than those targeting proteostasis. Crucially, they found that most drugs in PRISM do not significantly decrease cell viability in these cell lines, indicating that cells with high mutational loads are not broadly vulnerable to all classes of targeted therapies. Specifically, drugs inhibiting transcription, cytoskeleton organization, protein degradation, chaperones, and protein synthesis, and promoting apoptosis, were the most efficacious in targeting these cells. This further delineates additional drugs that may be effective against cancer cells with high mutational loads.

Figure 5 in the paper: classes of drugs resulting in increased cancer cell death with increased mutational load in cancer cell lines.
Conclusions + takeaways
This work is truly amazing and fundamental, initiating from a basic science question and producing translational insights that can be applied in the clinic. There are major implications of this paper:
1) Cancers with high mutation burdens may be effectively targeted via drugs that exacerbate protein misfolding stress, e.g., chaperone and proteasome inhibitors. The use of such drugs specifically against tumors with high mutational loads has not yet been explored clinically and should be. Protein misfolding stress may still yield additional vulnerabilities in highly mutated tumors waiting to be uncovered, with the possible addition of more classes of drugs to the toolbox in the future.
2) These results may suggest that the requirement to buffer protein misfolding stress could be a conserved hallmark of both cancer and normal somatic evolution in aging. The authors aptly stated that their results “contribute to an accumulating body of evidence that cancer and aging are different manifestations of related underlying evolutionary processes.” Indeed, disrupted proteostasis has been described as one of the hallmarks of aging in a seminal 2013 Cell paper [3]. Not only do highly mutated cancer cells have to compensate for this stress, but so do normal somatic aging cells, which also eventually accrue high mutational loads over longer periods of time. Despite this commonality between normal somatic tissues and tumors, aging cells appear to harness different defenses against proteostasis disruption, such as up-regulating chaperones in the Small HS family and autophagy, which were not leading responses seen in tumors in this study. This discrepancy needs to be further explored and investigated.
Final note + impressions
I want to note how impressive it is that such a study was performed using solely existing public datasets. Kudos to the authors! To glean such fundamental insights into cancer biology and then further extract clinically relevant information from existing datasets is a tremendous feat. That the identified therapeutic vulnerabilities may be cell-intrinsic and generic across cancer types is additionally exciting and promising. I look forward to seeing what comes of this!
This study is also of course fascinating from the perspective of cancer vs. aging. Aberrant peptide aggregation is a prevalent feature of (neuro)degenerative diseases, such as Alzheimer’s, Parkinson’s, Huntington’s, prion disease, and ALS [4]. Proteostasis disruption plays an indisputable role in aging; this study now shines light on its significant role in the cancer context, further strengthening the inextricable link between cancer and aging.
In hindsight, it makes a lot of inherent sense that cancer cells have to compensate for proteostasis disruption resulting from accelerated somatic mutation accumulation in their genomes. How they manage to survive in spite of this stress by up-regulating proteostasis machinery exemplifies how elusive yet vulnerable cancer is, and highlights the importance of studying its evolution.
References
Tilk, S., Frydman, J., Curtis, C., & Petrov, D. (2022). Cancers adapt to their mutational load by buffering protein misfolding stress. bioRxiv.
Tilk, S., Curtis, C., Petrov, D. A., & McFarland, C. D. (2021). Most cancers carry a substantial deleterious load due to Hill-Robertson interference. bioRxiv, 764340.
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194-1217.
Kikis, E. A., Gidalevitz, T., & Morimoto, R. I. (2010). Protein homeostasis in models of aging and age-related conformational disease. Protein Metabolism and Homeostasis in Aging, 138-159.